음전하를 띤 본질적으로 무질서한 영역은 DNA 결합 단백질에 의한 표적 검색을 가속화할 수 있습니다.

추상적인
진핵생물에서 많은 DNA/RNA 결합 단백질은 큰 음전하를 가진 본질적으로 무질서한 영역(IDR)을 가지고 있으며, 그 중 일부는 연속적인 서열의 아스파르테이트(D) 또는 글루타메이트(E) 잔기를 포함합니다. 이를 D/E 반복이라고 합니다. D/E 반복의 기능적 역할은 잘 알려져 있지 않지만 일부는 기능 영역과의 분자 내 정전기 상호 작용을 통해 자동 억제를 유발하는 것으로 알려져 있습니다. 이 연구에서 우리는 고 이동성 그룹 상자 1 (HMGB1) 단백질과 다양한 길이의 D / E 반복과 융합 된 Antp 호메오 도메인의 인공 단백질 구조에 대한 표적 DNA 검색 역학에 대한 D / E 반복의 영향을 조사했습니다. 우리의 실험 데이터는 특정 길이의 D/E 반복이 비기능성 고친화성 리간드('미끼')의 압도적인 존재에서 표적 연관성을 가속화할 수 있음을 보여주었습니다. 우리의 거친 입자 분자 역학 (CGMD) 시뮬레이션은자가 억제 된 단백질이 DNA에 결합하여 정전기적으로 구동되는 유도 맞춤 과정을 통해 DNA와 억제되지 않은 복합체로 전환 될 수 있음을 보여주었습니다. CGMD 시뮬레이션과 함께 우리의 운동 모델은 D/E 반복이 미끼가 있는 상태에서 표적 연관 프로세스를 가속화할 수 있는 방법을 설명할 수 있습니다. 이 연구는 대상 검색 프로세스에서 음전하를 띤 IDR의 전례 없는 역할을 조명합니다.

소개
진핵생물에서 전사 인자, 히스톤 및 기타 건축 단백질과 같은 대부분의 DNA 결합 단백질은 현저한 정도로 본질적으로 무질서한 영역(IDR)을 포함합니다(1). 예를 들어, 인간 전사 인자 중에서 IDR은 단백질 서열 길이의 평균 50%를 차지합니다(2). DNA 결합 단백질의 IDR은 단백질-단백질 상호 작용, 번역 후 변형 및 액체-액체 상 분리를 통해 중요한 조절 역할을 합니다(3,4). 폴더블 시퀀스와 비교하여 IDR 시퀀스는 극성 및 하전 아미노산이 풍부합니다(1). 어떤 경우에는 아스 파르 테이트 (D) 또는 글루타메이트 (E) 잔기 만 포함하는 고도로 음전하를 띤 세그먼트가 관찰됩니다 (5-20). 이를 'D/E 반복'이라고 합니다.
생물 정보학 연구에 따르면 D / E 반복은 진핵 생물의 단백질체에서 널리 퍼져 있습니다 (18,20). D / E 반복을 포함하는 단백질의 약 절반은 DNA / RNA 결합 단백질입니다 (20). D/E 반복의 구조적 유연성과 큰 음전하를 감안할 때, D/E 반복은 동일한 폴리펩티드 사슬 내에서 양전하를 띤 DNA-결합 도메인과 정전기적으로 상호작용할 수 있을 것으로 보인다. 이러한 상호작용은 DNA에 대한 명백한 친화도를 감소시키는 자가억제를 야기할 수 있다. 실제로 D/E 반복을 포함하는 음전하를 띤 IDR을 통한 자가 억제는 HMGB1, RFX1, Sox11 및 UBF1을 포함한 일부 DNA 결합 단백질에서 확인되었습니다(그림 1)(7–9,11,13). 그러나 D/E 반복에 의한 자동 억제의 역할은 잘 알려져 있지 않습니다.

인간 DNA 결합 단백질에서 D/E 반복의 예. D/E 반복에 의한 자동 억제는 HMGB1, RFX1, Sox11 및 UBF1(5,7–11,13)에 대해 실험적으로 나타났습니다.
자동 억제는 일반적으로 'on'과 'off'상태 사이의 분자 전환 측면에서 논의됩니다 (21-23). 어떤 경우에는 인산화와 같은 번역 후 변형에 의해 조절됩니다 (22,24-28). 단백질의 자가 억제 상태는 분자 스위치의 '꺼짐' 상태에 해당합니다. 그러나, 이러한 설명은 DNA-결합 단백질에 대해 단순할 수 있다. 많은 DNA 결합 단백질의 기능은 DNA의 표적에 결합해야 하지만 수많은 비기능적 고친화성 부위('미끼')가 단백질을 포획할 수 있습니다. 예를 들어, 특정 서열을 인식하는 전사 인자의 경우 게놈에는 단백질을 격리 할 수있는 수백만 개의 자연 미끼가 포함되어 있습니다 (29-31). 단백질이 억제되지 않은 상태로 유지되면 풍부한 미끼가 단백질을 쉽게 포획하여 표적과의 연관성을 방해 할 수 있습니다. 이러한 상황을 감안할 때, DNA 결합 단백질의 자가 억제는 격리의 위험을 감소시킬 수 있고, 따라서 미끼의 압도적인 존재 하에서 단백질-표적 연관 과정을 가속화할 수 있다.
이 논문에서 우리는 D/E 반복이 표적 DNA 검색의 가속화에 매우 적합하다는 것을 보여줍니다. D/E 반복의 영향을 조사하기 위해 고이동성 그룹 상자 1(HMGB1) 단백질과 다양한 길이의 D/E 반복 꼬리(DERT)와 융합된 Antp 동종 도메인의 인공 구조물을 사용했습니다. HMGB1은 C- 말단에 30 잔기 D / E 반복을 포함합니다. 이 단백질은 또한 시스플라틴 변형 DNA (32,33), 홀리데이 접합 (34,35), 부풀어 오른 DNA (36,37) 및 G- 쿼드러플 렉스 (38,39)와 같은 비정형 DNA를 인식하는 두 개의 DNA 결합 도메인을 포함합니다. HMGB1은 핵과 세포 외 공간에서 중요한 역할을하는 다기능 단백질입니다 (40,41). 핵에서 HMGB1은 DNA 샤페론 역할을 하며 전사 인자, DNA 복구/재조합 효소 및 염색질 리모델링 인자(40,42,43)를 돕습니다. 왜곡 된 DNA에 대한 신속한 접근은 HMGB1 (40)에 필수적입니다. HMGB1 자동 억제에 대한 이전 연구(13,44)를 기반으로 우리는 운동 실험을 수행했으며 D/E 반복이 압도적인 미끼 존재에서 HMGB1의 표적 검색 프로세스를 가속화한다는 것을 발견했습니다. Antp 호메오도메인의 조작된 단백질에 대한 데이터는 D/E 반복의 가속 효과가 다른 시스템에서 인위적으로 구현될 수 있음을 보여줍니다. 당사의 거친 분자 역학(CGMD) 시뮬레이션 및 운동 모델은 가속을 위한 메커니즘과 조건에 대한 훌륭한 통찰력을 제공합니다.
재료 및 방법
HMGB1 및 그 변형
인간 전장 HMGB1 및 이의 Δ30 변이체는 대장균 균주 BL21(DE3)에서 발현되었고, 우리의 이전 논문(13)에 기술된 바와 같이 양이온 교환, 음이온 교환 및 크기 배제 크로마토그래피 방법을 사용하여 정제되었다. 정제된 단백질을 동결건조하여 사용 전까지 -20°C에서 보관하였다. 모든 시스테인 잔기는 5 mM DTT에 의해 티올 형태로 환원되었고, 환원된 상태는 앞서 기술한 바와 같이 NMR에 의해 확인되었다.
Antp 호메오도메인 유도체
NdeI/EcoRI 부위에서 Antp HD-DERT49 단백질의 합성 유전자를 보유하는 pET16a 유도체 플라스미드는 GenScript에서 구입했습니다. Antp HD-DERT16을 대장균 균주 BL21(DE3)에서 발현시키고, SP-FF 양이온 교환, S-100 세크릴 크기 배제 및 리소스-Q 음이온 교환 컬럼을 통해 정제하였다. SP-FF 컬럼을 50 mM 포스페이트 완충액(pH 7.5) 및 0.1 M NaCl로 평형화시켰다. 단백질을 0.1-2.0 M NaCl의 구배로 용리시켰다. 단백질을 함유하는 분획을 ∼10 ml로 농축시키고, 50 mM Tris•HCl (pH 7.5), 1 mM EDTA, 및 0.4 M NaCl의 완충액으로 평형화시킨 크기 배제 크로마토그래피에 의해 추가로 정제하였다. Antp HD-DERT16 단백질을 함유하는 분획을 결합하고, 50 mM Tris•HCl (pH 7.5) 및 1 mM EDTA의 완충액으로 0배 희석하고, 자원-Q 음이온 교환 컬럼 상에 로딩하고, 2 mM Tris•HCl (pH 1.5) 및 50 mM EDTA에서 7.5-1.11 M NaCl의 구배로 용리시켰다. Antp HD–DERT7 단백질, Antp HD–DERT16 단백질 및 DERT가 없는 대조군 단백질의 대장균 발현을 위한 플라스미드는 QuikChange 라이트닝 키트(애질런트)를 사용한 돌연변이 유발을 통해 Antp HD-DERT16 단백질의 플라스미드에서 생성되었습니다. 이들 단백질은 Resource-Q 음이온 교환 컬럼 크로마토그래피가 0 mM Tris•HCl (pH 1.5) 및 50 mM EDTA에서 7–5.1 M NaCl의 구배를 갖는 Resource-S 양이온 교환으로 대체되었다는 점을 제외하고는 Antp HD-DERT280에 대해 상기 설명한 것과 본질적으로 동일한 방식으로 정제되었다. 개별 단백질의 농도는 ProtParam 도구 (https://web.expasy.org/protparam/)에 의해 예측 된 흡광 계수와 함께 <> nm에서의 UV 흡광도를 사용하여 측정되었습니다.
핵산
화학적으로 합성 된 DNA 가닥은 Integrated DNA Technologies에서 구입했습니다. 각 가닥을 음이온 교환 크로마토그래피로 정제하였다. DNA 이중체를 제조하기 위해 상보적인 가닥을 어닐링하고 음이온 교환 크로마토 그래피를 통해 과량의 단일 가닥 DNA를 제거했습니다. 이중 가닥 DNA의 농도는 Tataurov et al(260)의 방법을 사용하여 뉴클레오티드 서열로부터 계산된 흡광 계수와 함께 45nm에서의 UV 흡광도를 사용하여 측정되었습니다. 시스플라틴 변형을 갖는 형광 표지된 20-bp DNA를 앞서 기술한 바와 같이 제조하였다(13). 효모 tRNA는 시그마-알드리치(cat#10109517001)로부터 구입하였다. tRNA 농도를 측정하기 위해 260nm에서의 UV 흡광도를 7 × 10의 흡광 계수와 함께 사용했습니다.5 M−1센티미터−1, 평균 길이와 저색성 (46,47)에서 추정되었습니다.
시약
전술한 물질을 제조하고 아래에 설명된 실험을 수행하기 위해 사용된 화학물질은 달리 명시되지 않는 한 Sigma Aldrich로부터 구입하였다.
결합 친화도 측정
Antp HD-DERT 구축물의 결합 친화도는 이전에 기술된 바와 같이 TAMRA 표지된 15-bp DNA 듀플렉스 및 다양한 농도의 단백질을 사용하는 형광 이방성 기반 단백질 적정 실험을 통해 측정되었습니다(48). 미끼 15-bp DNA의 친화도는 이전에 기술된 바와 같이 형광 이방성 기반 경쟁적 결합 분석을 통해 측정되었습니다(48). 효모 tRNA 혼합물에 대한 HMGB1의 명백한 친화도는 시스플라틴 변형, 4nM HMGB40 및 다양한 양의 tRNA (1-10,10nM)를 포함하는 000nM FAM 표지 DNA 듀플렉스를 사용하는 형광-이방성 기반 경쟁 결합 분석을 통해 측정되었습니다. HMGB1•tRNA 복합체에 대한 겉보기 해리 상수 Kd는 시스플라틴-변형-DNA(2)을 갖는 HMGB48 복합체에 대한 Kd 및 수학식 1의 수학식 13와 함께 측정된 FAM 형광 이방성 데이터로부터 결정되었다.
미끼가 있는 상태에서 표적 검색 역학 측정
미끼가 있는 상태에서 표적 연관 역학은 응용 광물리학 SX25-LED 정지 흐름 분광형광계를 사용하여 20°C에서 측정되었습니다. 470nm에서 최대 강도를 갖는 편광 LED 광을 FAM 형광단의 여기(excion)에 사용하였다. 형광 이방성은 편광판이 있는 T-형식 구성으로 배치된 두 개의 방출 채널과 각각에 대해 515nm에서 컷오프가 있는 장통과 필터를 사용하여 실시간으로 측정되었습니다. 모든 결합 반응은 Ttot ≪ Ptot ≪ Dtot의 조건 하에서 수행되었으며, 여기서 Ttot, Ptot 및 Dtot는 각각 표적 (즉, 프로브), 단백질 및 디코이의 총 농도이다. 표적 연관성에 대한 명백한 의사 8차 운동 속도 상수(kapp)는 형광 이방성의 시간 과정으로부터 단일-지수 피팅을 통해 결정되었습니다. 속도 상수 kapp는 단백질의 다양한 농도에서 측정되었다. 각 운동 속도 상수에 대해, 측정은 10-1 회 반복되었다. MATLAB 소프트웨어(MathWorks)는 비선형 최소제곱 피팅에 사용되었습니다. HMGB30 및 그 Δ1 변이체에 대한 정지 흐름 실험을 위해, 다음 두 가지 용액을 정지 흐름 장치에 의해 1:80 부피비(각각 10μl)로 빠르게 혼합했습니다: 단백질 용액 및 시스플라틴 변형 및 20nM tRNA를 포함하는 8000nM FAM 표지된 10-bp DNA의 DNA/RNA 용액을 미끼로 사용합니다. 두 용액 모두 7 mM 인산칼륨 (pH 5.1), 1 mM DTT, 2 mM MgCl100 및 11 mM KCl을 함유하는 완충제 내에 있었다. 혼합을 위한 흐름이 중단된 직후, 형광 이방성의 시간 코스 데이터를 0.02 내지 0.05초 범위의 시간 간격으로 33초의 기간 동안 수집하였다. Antp HD 유도체에 대한 정지 흐름 실험을 위해, Antp 인식 서열 (10 nM)을 포함하는 FAM 표지 된 15-bp DNA를 형광 표지 표적으로, 4000-bp DNA 듀플렉스 (33 nM)를 미끼로 사용하였다. 3-bp DNA의 서열은 FAM-AGCCATTACAGTACGCACGTACGGTGGCACGA-15'이었고, 여기서 Antp 인식 서열에 밑줄이 그어져 있다. 비특이적 10-bp DNA의 서열은 AGAAAGCAGACAGAG였다. 완충액은 7 mM 인산칼륨 (pH 5.100) 및 10 mM KCl이었다. 형광 표지 표적과 Antp- 유도체 단백질의 연관성의 과정은 0.001-0.05 초의 시간 간격으로 <> 초 동안 형광 이방성의 시간 경과 데이터를 통해 분석되었다.
NMR 실험
NMR 실험은 TCI 극저온 프로브가 장착된 브루커 아반스 III 800MHz NMR 분광기를 사용하여 수행하였다. C-단자 D/E 반복과 Antp HD의 정전기 상호작용을 조사하기 위해 1H–15N TROSY 스펙트럼은 ∼25.0–1.0 mM 동안 2 ° C에서 기록되었습니다. 15N-표지된 Antp HD-DERT11 및 상응하는 단백질에 대해 11, 200, 300, 400, 500 및 700 mM KCl에서 DERT900 (Antp HD-MID)이 결여되었다. 더 얇은 내부 튜브에 단백질 용액이 있고 외부 튜브에D2O가 있는 동축 NMR 튜브를 사용하여 저온 프로브의 임피던스 정합을 최적화하는 문제를 회피했습니다. 1높은 이온 세기로 인한 RF 회로(49). 단백질 용액은 또한 NMR 화학적 이동 참조(10)를 위한 7mM 인산칼륨(pH 5.1) 및 2mM 2,2-디메틸-5-실라펜탄-50-술포네이트(DSS)를 함유하였다. NMR 스펙트럼은 NMR-Pipe (51) 및 NMRFAM-SPARKY (52) 프로그램에 의해 처리 및 분석되었다. Antp HD에 대한 이전 NMR 연구(53)로부터의 호메오도메인 내의 백본 NH 그룹에 대한 공명 할당이 분석에 사용되었다. 이전 연구(13)에서 설명한 접근 방식을 사용하여 평형 상수 Kai(= [X]/[P]) 및 자가 억제 상태의 집단은 Antp HD-DERT11과 Antp HD-MID 단백질 사이의 화학적 이동 차이(Δδ)의 염 의존성 데이터로부터 결정되었습니다.
전산 모델링
모든 원자 시뮬레이션
92-잔기 Antp HD-DERT 단백질을 구성하기 위해 NMR 용액 구조(PDB: 1HOA)의 모델 20(2개 중)을 사용했는데, 이 구조의 코어 형태는 DNA에 결합된 Antp HD의 결정 구조와 매우 유사하고 말단이 확장되었습니다. NMR 구조는 Antp HD의 60개 잔기 모두에 대한 좌표를 제공하는 반면 X선 결정 구조에는 일부 N- 및 C-말단 잔기가 없기 때문에 사용되었습니다. 단백질의 p53 세그먼트 (15개 잔기)를 NMR 구조 (PDB ID 2K8F), 사슬 B로부터 취하고, 모델 1 (10개 중)을 사용하였고, 여기서 세그먼트는 나선형이었다. 최종 16 개의 산성 잔류 물은 PyMOL을 사용하여 나선형 형태로 구축되었습니다. 조각들은 COOT를 사용하여 함께 도킹되었다. GROMACS(v. 2019.3)를 사용하여 유리 Antp HD-DERT 단백질 및 DNA와의 복합체에 대한 모든 원자 시뮬레이션을 실행했습니다. 단백질, SPC 물 및 이온에 대한 포스장 파라미터는 AMBER99SB-ILDN 포스 필드로부터 유도되었다. 모든 구조물을 십이면체 상자에 넣고 용매화시켰다. 나트륨 및 염화물 이온을 0.125M의 농도로 첨가하고 전체 전하를 중화하기 위해 약간 조정했습니다. 모든 구조는 최소화 및 NVT 및 NPT 평형화를 거친 후 각각 3000ns의 <>회 생산 실행을 거쳤습니다.
거친 분자 역학 시뮬레이션
Antp HD-DERT 유도체의 역학과 DNA에 대한 결합은 거친 입자 분자 역학(CGMD) 시뮬레이션을 사용하여 연구되었습니다. 각각의 잔기는 그의 Cα 원자의 위치에서 단일 비드로 표시되었다. DNA는 인산염, 당 및 염기를 나타내는 뉴클레오티드 당 54 개의 비드로 모델링되었으며 각 비드는 그룹의 기하학적 중심에 위치했습니다. 시뮬레이션에 적용된 힘장은 네이티브 접촉을 보상하는 Lennard-Jones 잠재력과 비 네이티브 접촉에 불이익을 줄 수있는 반발 잠재력을 포함하는 네이티브 토폴로지 기반 모델을 사용했습니다 (56–100). DNA는 시뮬레이션 내내 제자리에 있고 단단하게 유지되는 1개의 염기쌍 길이를 가진 선형 이중 가닥 B-DNA 분자로 모델링되었습니다. 단백질의 양전하를 띤 잔기(Lys, Arg)에는 (+1e)의 점 전하가 할당되었고, 음전하를 띤 잔기(Asp, Glu)와 DNA 백본의 인산염 비드에는 음전하(-57e)가 할당되었습니다. 하전된 비드 qi,qj 사이의 정전기 전위는 Debye-Hückel 상호작용에 의해 모델링되었으며, 이는 수용액에 침지된 용질의 이온 강도를 설명합니다(58). 힘장의 명시적 형태는 다른 곳에 보고된다(9). Antp HD의 구조는 결정 구조 PDB 59ANT의 형태를 기반으로했습니다. 우리는 DERT에서 음전하를 띤 잔류물의 수가 다른 Antp HD-DERT의 전산 설계 변형을 가지고 있습니다. DERT를 돌연변이시키는 것은 길이를 고정시키고 N- 말단에 위치한 전하를 중화시킴으로써 달성되었습니다. CGMD에서 D/E 반복과 단백질의 다른 부분 간의 상호 작용은 정전기 상호 작용에 의해서만 모델링되었습니다. 단백질 내의 다른 모든 상호작용은 CSU 프로그램을 사용하여 원래 구조에서 접촉이 정의되지 않는 한(즉, 제외된 부피)로 모델링되었다(60). 분리 및 DNA의 존재 하에서 단백질의 역학은 Langevin 방정식 (80)을 사용하여 시뮬레이션되었다. 유전 상수는 80이었고 소금 농도는 본문 전체에서 언급 한대로 다양했습니다. 각 시스템에 대해 2 × 10으로 구성된 최소 <> 개의 시뮬레이션을 수행했습니다.8 MD 단계. 궤적 프레임은 1000 단계마다 저장되었습니다.
방정식 기반 시뮬레이션 평가
운동 모델에 대한 속도 방정식은 보충 데이터에 나와 있습니다. 개별 종의 농도의 시간 과정은 속도 방정식을 수치로 풀어서 계산되었습니다. 운동 시뮬레이션은 'ode15s' 경직성 상미분 방정식 솔버를 사용하여 MATLAB 스크립트로 수행되었습니다. 겉보기 속도 상수 kapp는 단백질-표적 복합체 형성의 시뮬레이션된 시간 경과에 대한 단일지수적 피팅([PT]/Ttot)에 의해 결정되었습니다. 개별 종의 평형 농도는 평형 상수와 질량 보존에 대한 동시 방정식을 풀어 'solve'함수를 사용하여 MATLAB 스크립트로 계산되었습니다.
통계 분석
운동 측정의 경우 평균값을 계산하기 위해 최소 5번의 반복실험이 사용되었습니다. 친화성 측정은 95배로 증가했습니다. 보고된 실험 값은 평균의 평균 및 표준 오차(SEM)입니다. 피팅 모수의 경우 오차 막대는 <>%의 신뢰 구간을 나타냅니다.
결과
D/E 반복에 의한 자동 억제는 HMGB1-표적 연관성을 가속화합니다.
HMGB1은 동적 자가억제를 거치는 다기능 DNA 결합 단백질입니다. 세포핵에서 HMGB1은 구조적으로 왜곡된 DNA에 결합하여 DNA 샤페론으로 작용합니다(40,43) HMGB1은 시스플라틴 변형 DNA, 34방향 접합 DNA 및 G-쿼드러플렉스(39,61,62)와 같은 다양한 비B형 DNA에 대해 강한 친화력을 나타냅니다. 또한 분지형 RNA에 결합할 수 있다(30). HMGB1의 C-말단 2 잔기는 D/E 반복이다(그림 9A). 이 음전하를 띤 세그먼트는 두 개의 DNA 결합 도메인 및 동일한 분자 내의 다른 양전하를 띤 영역과의 정전기 퍼지 상호 작용을 통해 동적 자동 억제를 유발합니다 (13,1). HMGB10 자동 억제에 대한 평형 상수 Kai는 염 농도에 크게 의존하며 ∼10-<> 정도입니다.2 생리적 이온 강도 (13).

HMGB1의 자동 억제는 미끼가있는 상태에서 HMGB1- 표적 연결을 가속화합니다. (A) HMGB1의 자가 억제는 두 개의 DNA 결합 도메인 및 기타 양전하를 띤 영역과 D/E 반복의 정전기적 상호 작용을 통해 발생합니다(8-10). D/E 반복이 없기 때문에 Δ30 변이체는 자가 억제를 거치지 않습니다. 시스플라틴에 의해 변형된 형광 표지된 프로브 DNA가 또한 도시되어 있다. (B) 미끼의 존재 하에서 단백질-표적 결합의 동역학 조사를 위한 정지-흐름 형광 실험. (c) 단백질 용액과 프로브 DNA 및 tRNA를 미끼로 포함하는 DNA 용액의 혼합시 FAM 형광 이방성의 시간 경과. 단백질, 프로브 DNA 및 tRNA의 농도는 각각 50, 10 및 8000 nM이었다. (d) 미끼로서 8 μM tRNAs의 존재 하에서의 단백질-표적 회합에 대한 명백한 의사 10차 속도 상수kapp의 단백질 농도 의존성. 완충액은 7 mM 인산칼륨 (pH 5.1), 1 mM DTT, 2 mMMgCl100 및 <> mM KCl이었다.
HMGB1의 표적 DNA 검색에 대한 D/E 반복의 영향을 조사하기 위해 전장 HMGB1 단백질의 거동을 Δ30 변이체의 거동과 비교했습니다. 30-잔기 D/E 반복 매개 자가억제의 부족으로 인해, 시스플라틴 변형 DNA에 대한 Δ30 변이체의 친화도는 이전에 입증된 바와 같이 생리학적 이온 강도에서 전장 단백질보다 >∼100배 더 강합니다(13). 정지 흐름 형광 실험을 통해 미끼가 있는 상태에서 전장 HMGB1 단백질과 Δ30 변이체에 대한 단백질-표적 연관 역학을 측정했습니다(그림 2B). FAM-표지된 시스플라틴-변형된 DNA (10 nM; 20 염기쌍 [bp])를 표적으로서 사용하였다. tRNA(8000 nM)를 미끼로 사용하였다. 우리는 분지 RNA가 HMGB1에 효과적으로 결합 할 수 있고 (62) RNA가 핵에 매우 풍부하여 생체 내에서 HMGB1을 가두는 미끼 역할을 할 수 있기 때문에 선형 DNA 이중체보다는 tRNA를 선택했습니다. 그림과 같이 보충 그림 S1 보충 데이터에서 tRNA에 대한 HMGB1의 친화도는 시스플라틴 변형 DNA에 대한 것보다 ∼500배 더 약합니다. 정지 흐름 실험에서, 단백질 용액을 프로브 DNA 및 tRNA를 포함하는 용액과 혼합한 후 FAM 형광 이방성을 모니터링하였다. 시간 경과 데이터로부터, 우리는 풍부한 미끼가있는 단백질의 다양한 농도에서 표적 연관성에 대한 겉보기 속도 상수를 결정했다.
정지 유동 동역학 데이터의 일부 예가 도 2C에 도시되어 있다. 이들 실험에서, 전장 HMGB1 단백질은 Δ30 변이체보다 현저하게 더 빠른 표적 연관성을 나타내었지만, 자가억제는 전장 HMGB1의 친화도를 Δ30 변이체보다 약하게 만든다. 단백질 농도가 50nM일 때, HMGB1의 표적 회합은 자가억제가 결여된 Δ10 변이체의 표적 회합보다 30배 빨랐다. 미끼의 존재 하에서의 표적 회합 동역학의 단백질 농도 의존성은 일반적으로 비선형이지만, 단백질 농도가 미끼 농도보다 훨씬 낮을 때 사실상 선형일 것으로 예상된다(63). 실제로, 두 단백질 모두 테스트된 단백질 농도 범위에 대해 거의 선형 의존성을 나타냈습니다(그림 2D). 각 농도에서, 전장 HMGB1 단백질은 Δ30 변이체보다 현저하게 더 빠른 회합을 나타내었다. 이러한 데이터는 D/E 반복을 통한 자동 억제가 미끼가 있는 상태에서 HMGB1-표적 연관성을 가속화한다는 것을 시사합니다.
D/E 반복 꼬리를 이용한 인공 자가억제 시스템
D/E 반복이 표적 검색 역학의 가속화를 유발하는지 여부를 추가로 조사하기 위해 링커를 통해 D/E 반복과 연결된 Antp 항상성 도메인(HD, 전체 전하, +12e)의 3가지 단백질 구조를 만들었습니다(그림 64A). Antp HD는 TAATG 서열을 인식하는 DNA 결합 도메인입니다 (53). Antp HD 구조에는 D/E 반복 꼬리(DERT)가 부착되었으며 세린 또는 알라닌 잔기로 대체하여 길이를 다양화했습니다. HD와 DERT 사이의 링커 서열은 본질적으로 무질서하지만 일부 (∼15 %) 나선형 성향을 갖는 p29 잔기 30-65로부터 채택되었다 (15). 형광 이방성 기반 분석을 통해 Antp 인식 서열을 포함하는 3-bp DNA에 대한 친화도를 측정하고 DERT가 각 단백질 구축물에 대해 자가억제를 유발한다는 것을 확인했습니다(그림 <>B; 참조 보충 그림 S2 보충 데이터에서).
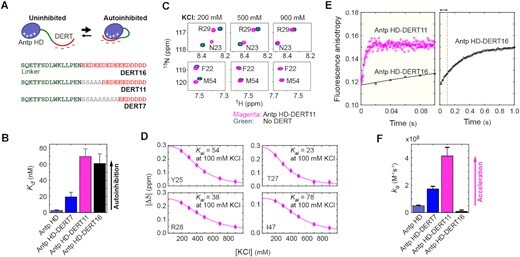
D/E 반복 꼬리(DERT)를 사용하는 인공 자가억제 시스템. (a) DERT (적색)가 부착된 Antp 호메오도메인의 단백질 작제물. 녹색으로 표시된 서열은 p53 잔기 15-29로부터 채택된 링커이다. (b) Antp 인식 서열을 포함하는 15-bp DNA와 단백질 구축물의 복합체에 대한 해리 상수 (Kd). (C) 중첩 된 이종 핵 1H–15다양한 농도의 KCl에서 Antp HD-DERT11 및 DERT가 없는 대조군 단백질에 대해 기록된 N 상관 스펙트럼. HD와 DERT 사이의 분자 내 정전기 상호 작용으로 인해 두 구조의 NMR 화학적 이동은 낮은 이온 강도에서 크게 다릅니다. (D) Antp HD-DERT11과 DERT가없는 대조군 단백질 간의 화학적 이동 차이. 100 mM KCl에서의 자가억제 평형 상수 Kai는 앞서 기술한 바와 같이 각 잔기에 대한 화학적 이동의 KCl 농도 의존성에 대한 피팅을 통해 결정되었다(13). 실선 곡선은 최적 적합 곡선을 나타냅니다. 상이한 잔기 사이의 Kai 상수의 변화는 자가억제 상태에서의 DERT의 동적 성질을 반영할 수 있다 (13). (E) 200 nM 단백질을 10 mM KCl에서 33 nM 4-bp FAM 표지 DNA (Antp 인식 서열 포함) 및 15 μM 100-bp 미끼 DNA의 용액과 혼합 할 때 측정 된 정지 흐름 형광 이방성 데이터. Antp HD-DERT11은 매우 빠른 동역학을 나타냈기 때문에 이방성 측정 사이의 시간 간격은 이 단백질에 대해 더 작은 값으로 설정되었습니다. 더 큰 소음은 더 짧은 시간 간격 때문입니다. (f) 겉보기 회합 속도 상수ka는 정지 유동 형광 동역학 실험으로부터의 단백질 농도 의존성 데이터를 사용하여 결정된다.
통해 1H–15N개의 이종핵 NMR 실험에서, 우리는 HD와 DERT 영역 사이의 상호작용을 조사하였다. 우리의 이전 연구는 전장 HMGB1 단백질과 Δ30 변이체 사이의 NMR 화학적 이동의 차이가 D / E 반복을 통한 자동 억제의 정전기 특성으로 인해 이온 강도에 크게 의존한다는 것을 보여주었습니다 (13). 마찬가지로, NMR 화학적 이동은 낮은 이온 강도에서 DERT가 있는 구조물과 없는 구조물 간에 상당히 달랐습니다(그림 3C). 이온 강도가 증가함에 따라 화학적 이동 차이가 감소하여 3에 가까워졌습니다(그림 13D). 이러한 결과는 DERT가 인공 자가억제 구축물에서 양전하를 띤 HD와 정전기적으로 상호 작용한다는 것을 강력하게 시사합니다. 우리는 빠른 교환에서 자동 억제 및 억제되지 않은 상태를 반영하는 명백한 NMR 화학적 이동을 분석했습니다. 앞서 설명한 바와 같이(도 11), NMR 화학적 이동 차이의 이온-강도 의존성은 반대이온 축합 이론에 기초한 모델 함수에 적합하였다. 피팅으로부터의 100 mM KCl에서의 Antp HD-DERT3 구축물에 대한 자가억제 평형 상수 Kai를 도 <>D에 나타내었다.
각 인공 자가 억제 단백질에 대해 정지 흐름 형광 실험을 수행하여 미끼가 있는 상태에서 표적 연관 역학에 대한 DERT의 영향을 조사했습니다. 이 경우, Antp 인식 서열을 함유하는 FAM-표지된 33-bp DNA 듀플렉스가 표적으로서 사용된 반면, 표지되지 않은 비특이적 15-bp DNA 듀플렉스가 미끼로서 사용되었다. 우리는 단백질 용액을 0.01 μM 표적 및 4 μM 미끼를 포함하는 용액과 혼합한 직후 실시간으로 FAM 형광 이방성을 모니터링했습니다. 시간 과정의 예가 도 3E에 도시되어 있고, 단백질 농도 의존성으로부터 결정된 겉보기 회합 속도 상수가 도 3F에 도시되어 있다.
흥미롭게도, 단백질-표적 회합에 대한 자가억제의 동역학적 영향은 DERT 길이에 크게 의존하였다. Antp HD-DERT11 및 -DERT7 구축물은 표적 회합의 현저한 가속을 나타내었다. DERT7 구축물에 대한 더 작은 효과는 Kd 데이터에 반영된 바와 같이 더 약한 자가억제에 의해 설명될 수 있다. DERT16 및 DERT11 구축물이 표적에 대한 친화도 측면에서 유사한 정도의 자가억제를 나타내었지만, Antp HD-DERT16은 어떠한 가속도 나타내지 않았다; 오히려 대상 연결은 원래 Antp HD보다 훨씬 느렸습니다 (그림 3F). 우리의 추가 계산 조사 및 운동 모델은 Antp HD-DERT16의 이러한 다양한 동작에 대한 통찰력을 제공할 것입니다. 그럼에도 불구하고 Antp HD-DERT에 대한 우리의 현재 데이터는 단백질-표적 결합의 자가 억제 보조 가속이 단백질 공학을 통해 인위적으로 구현될 수 있음을 분명히 시사합니다.
거친 분자 역학 시뮬레이션에서 표적 검색 프로세스
표적 검색 과정에서 단백질은 해리 및 DNA와의 연관성 외에도 슬라이딩, 호핑 및 세그먼트 간 전달 과정을 거칩니다 (60,66,67). D/E 반복이 있는 DNA 결합 단백질은 검색 시 자가 억제 형태를 채택하고 표적 부위에서 억제되지 않은 형태로 변경할 수 있습니다. 이러한 메커니즘은 DNA 결합 단백질이 미끼에서 빠져 나와 표적을 찾는 데 도움이됩니다. 거친 분자 역학 (CGMD) 시뮬레이션은 표적 DNA 검색 과정에서 구조적 역학에 대한 실험 데이터를 해석하는 데 적합합니다 (68,69). 따라서 자가 억제 단백질이 DNA의 표적에 도달하고 억제되지 않은 상태로 전환하는 방법에 대한 추가 통찰력을 얻기 위해 Antp HD-DERT 단백질에 대한 CGMD 시뮬레이션을 수행했습니다.
먼저 CGMD를 적용하여 자동 억제 상태를 특성화했습니다. 시뮬레이션은 DERT가 N 말단 꼬리, 나선 3 (DNA 인식 나선) 및 나선 1과 2를 연결하는 루프와 동적으로 상호 작용한다는 것을 보여주었습니다. 구조적 앙상블은 높은 가소성으로 인해 광범위합니다. 따라서, DERT를 수반하는 모든 분자내 상호작용이 각각의 구조에서 동시에 발견되는 것은 아니다. CGMD 시뮬레이션에서 확인된 자동 억제 상태의 유사한 구조적 특징이 원자 시뮬레이션에서도 발견되었습니다(보충 데이터의 보충 그림 S3), CGMD의 유효성을 뒷받침합니다. DERT의 전하 함량이 DNA 결합 역학에 미치는 영향을 해결하기 위해 Antp HD-DERT의 다양한 구조를 사용하고 CGMD 시뮬레이션에서 선형 100bp DNA의 중간에 모델링된 표적 부위와의 연관성에 대한 비율을 계산했습니다. 동일한 DNA의 다른 부위도 Antp HD와 정전기적으로 상호 작용하고 미끼 역할을 합니다. 따라서, CGMD는 본질적으로 표적 및 미끼를 포함하는 단일 DNA 세그먼트의 분해능에서 표적 회합 동역학에서 자가억제의 역할을 조사하기 위한 유용한 도구를 제공한다.
CGMD 궤적을 사용하여 표적에 대한 단백질의 초기 결합 속도를 조사했습니다. 초기 결합이 반드시 표적과의 억제되지 않은 복합체로 이어지지 않을 수 있음에 유의한다. 우리의 데이터는 DERT에서 음전하를 띤 잔기가 더 많은 단백질의 경우 초기 결합 속도가 더 크다는 것을 보여주었습니다 (그림 4A, 검은 색 원). DERT의 음전하가 증가하면 자가억제 상태의 개체군이 증가하기 때문에(도 4B), 더 빠른 초기 결합은 자가억제 상태에서의 더 빠른 확산으로부터 비롯될 수 있다. 실제로, 도 4C(좌측 수직축)는 DERT 전하 함량의 증가에 따라 확산이 더 빨라진다는 것을 도시한다. 그림 4C(오른쪽 수직 축)는 또한 DERT가 더 충전됨에 따라 슬라이딩 모드에서 호핑 모드로의 전환을 보여줍니다. 슬라이딩은 DNA와의 접촉을 유지하면서 DNA를 따라 단백질 분자의 1D 확산입니다. 호핑은 해리, 3D 확산을 통한 유리 단백질의 짧은 여행, DNA의 근위 부위와의 재결합을 포함하는 과정입니다. 호핑으로의 전환은 자가억제 상태에서 단백질에 의한 표적 검색을 가속화할 수 있는 전반적으로 더 빠른 선형 확산을 초래할 수 있습니다.

D/E 반복을 통해 동적 자가 억제를 겪는 단백질에 의한 DNA 인식의 전산 분석. (A) 거친 시뮬레이션에서 계산 된 Antp HD-DERT의 동역학. 검은색 원: 단백질 분자가 DNA의 표적에 처음 결합하는 속도 상수. 이 결합 이벤트는 자동 억제 (k XT에 의해 측정) 또는 억제되지 않은 (kPT에 의해 측정) 상태로서 발생할 수있다. 흰색 원: 단백질이 표적에 있는 동안 자가억제(XT)에서 비억제(PT) 상태로의 전환에 대한 속도 상수 kXTPT. 속도 상수는 DERT에서 음전하를 띤 잔류물 수의 함수로 표시됩니다. (B) DERT에 대한 전하 수의 함수로서 Antp-HD DERT가 자동 억제 상태(상태 X)에 있을 확률. 이 데이터는 DNA가없는 상태에서 얻은 것입니다. (C) 왼쪽 y축(검은색 기호): DERT에서 하전된 잔기 수의 함수로서 DNA를 따라 선형 확산에 대한 확산 계수. 오른쪽 y축(회색 기호): 평행 이동과 회전 사이의 커플링 기울기입니다. -0.18의 기울기는 슬라이딩을 나타냅니다. (83) 확산 계수와 기울기에 대한 자세한 내용은 보충 데이터에 설명되어 있습니다(또한 참조 보충 그림 S4). (d) 각 Antp 잔기와 가장 가까운 DNA 포스페이트 사이의 최단 거리는 자가 억제(XT, 회색) 및 억제되지 않은 상태에 대해 표시됩니다. (PT, CGMD (검은 색 실선) 및 원자 시뮬레이션 (검은 색 점선)에서 계산 됨). y축은 DNA에 결합된 Antp의 결정 구조에서의 거리와의 차이를 나타낸다. (E) DERT와 Antp-HD 사이의 거리 (y 축) 대 Antp-HD와 DNA 사이의 거리 (x 축)를 보여주는 3 차원지도. 각 패널에는 그림에 표시된 대로 다른 변형에 대한 맵이 표시됩니다. 금지되지 않음(PT) 및 자동 금지(XT) 상태가 맵에 표시됩니다. 색상은 100차원 공간을 따라 투영된 Antp HD-DERT 변이체 형태의 모집단 확률(로그 스케일)에 해당합니다. (F, G) 자동 억제 (패널 F, XT) 또는 억제되지 않은 (패널 G, PT) 상태에서 표적에서 Antp HD-DERT의 선택된 형태. DERT는 빨간색으로 표시되고 나선 1은 파란색으로 표시됩니다. 선형 <>-bp DNA의 중간에 위치한 표적 부위는 빨간색으로 강조 표시됩니다. XT에서 PT로의 전환의 예는 보충 데이터의 영화 S<>에 나와 있습니다.
평균 통과 시간의 통계 분석을 통해 CGMD 궤적에서 자가 억제 복합체에서 억제되지 않은 복합체로 전환하기 위한 속도 상수 kXTPT를 계산했습니다. kXTPT 데이터는 DERT에서 하전 된 잔기의 수를 늘리면 전이가 느려지는 것을 나타냅니다 (그림 4A, 흰색 원). 우리의 계산 결과는 그림 4A의 두 곡선이 교차 할 때, 즉 HD-DERT에 ∼ 9 개의 대전 된 잔기가있을 때 특정 표적 인식의 최대 가속이 달성 될 것임을 시사합니다. 이 추세는 그림 3F에 표시된 실험 데이터와 매우 유사합니다.
자가 억제가 표적 연관 역학에 미치는 영향에 대한 추가 통찰력을 얻기 위해 시뮬레이션에서 Antp HD의 각 잔기와 가장 가까운 DNA 인산염 사이의 평균 거리를 계산했습니다. 억제되지 않은 상태에 대해 계산된 거리는 원자 및 CGMD 시뮬레이션 모두에서 DNA에 결합된 Antp-HD의 결정 구조에서 발견되는 거리에 매우 가깝지만(그림 4D, 점선 및 단색 검은색 곡선), 자동 억제 상태에서 나선 2와 3은 DNA에서 ∼1nm 멀리 이동합니다(그림 4D, 회색 곡선). 그림 4F–G의 스냅샷은 자동 억제 상태에서 DERT가 나선 2와 3을 결합하여 HD가 목표에 완전히 도달하는 것을 방지한다는 것을 분명히 보여줍니다. 대조적으로, 억제되지 않은 상태에서 DERT는 확장 된 형태에 있으므로 나선 3이 표적 부위와의 특정 상호 작용을 매개하도록 허용합니다. 영화 S1 보충 데이터 쇼 CGMD 궤적에서 자동 억제 복합체에서 억제되지 않은 복합체로의 전환의 전형적인 예입니다. 이러한 전이는 DNA와 DERT 사이의 정전기 반발력에 의해 촉진되는 것으로 보이며, 둘 다 강하게 음전하를 띠고 있습니다.
대상에서 자동 억제 상태와 억제되지 않은 상태 간의 전환을 더 잘 이해하기 위해 시뮬레이션에서 관찰된 전환의 에너지 환경을 분석했습니다. 그림 4E는 DERT의 질량 중심 (COM)과 HD의 구형 부분 (y 축) 사이의 거리와 HD의 COM과 DNA 축 (x 축) 사이의 거리를 사용하여 플롯 된 에너지 풍경을 보여줍니다. 자가 억제 복합체에서는 DERT와 HD 사이의 거리가 짧고 HD와 DNA 사이의 거리가 길어질 것으로 예상하며, 억제되지 않은 복합체의 경우 그 반대의 경우도 마찬가지입니다. 실제로, DERT의 전하가 작을 때, 단백질은 대부분 억제되지 않은 상태로 채워지고, 자동 억제는 드물다. DERT가 더 많이 충전됨에 따라, 자가억제 복합체의 개체군과 자가억제된 복합체와 억제되지 않은 복합체 사이의 에너지 장벽이 점차 증가하며, 이는 kXTPT의 감소에 따른 것이다.
D/E 반복의 영향을 설명하는 키네틱 모델
D/E 반복에 의한 표적 검색 역학의 가속화는 운동 모델을 사용하여 설명할 수 있습니다. 가속에 대한 메커니즘의 정성적 개념이 도 5A에 도시되어 있다. 유리 단백질은 억제되지 않은 상태 (P)와자가 억제 된 상태 (X) 사이에서 동적 평형을 겪습니다. 억제되지 않은 상태는 표적에 더 강한 결합을 허용하지만 단백질을 미끼에 갇힐 위험이 높습니다. 대조적으로, 자가 억제 상태는 결합 친화도를 감소시키지만 단백질이 미끼에 갇힐 위험을 낮춥니다. 이 두 상태를 동적 평형 상태로 유지함으로써 단백질은 표적에 더 빠르게 결합할 수 있습니다.
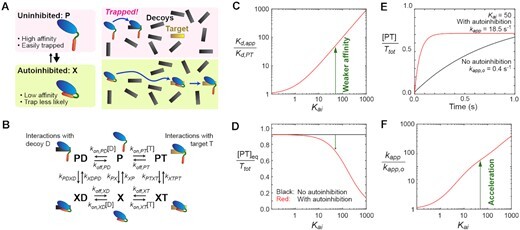
D/E 반복에 의한 자동 억제가 압도적인 미끼 존재 시 표적 검색을 가속화할 수 있는 방법을 설명하는 키네틱 모델입니다. (A) 자동 억제는 미끼에 갇힐 위험을 줄일 수 있습니다. (B) 표적 및 미끼가있는 상태에서 동적 자동 억제의 운동 모델. (c) 자가억제는 표적에 대한 겉보기 친화도를 감소시킨다. Kd,app는 미끼가 없는 경우의 단백질-표적 복합체에 대한 겉보기 해리 상수를 나타낸다. Kd,PT는 PT에 대한 고유 해리 상수이다. Kai는 자가억제에 대한 평형 상수를 나타낸다(Kai = [X]eq/[P]eq). (D) 미끼의 존재 하에서의 단백질 결합 표적의 개체군. 자가 억제 시스템에서 단백질-표적 복합체 (PT)의 평형 집단은 자동 억제가 미끼에 의한 단백질의 격리를 약화시키기 때문에 억제가없는 시스템에서보다 약간만 작을 수 있습니다. (E) 속도 방정식을 풀어 얻은 단백질-표적 복합체(PT)의 형성의 시간 과정. 초기 조건은 [D] = D tot, [T] = T tot, [P] = P tot/(1 + K ai), [X] = P tot K ai/(1 + K ai), [PT] = [PD] = [XD] = [XT] = 0이었다. 빨간색 곡선은 패널 B에 표시된 키네틱 모델에 대한 결과인 반면, 검은색 곡선은 자동 억제가 없는 시스템(즉, X, XD 및 XT가 존재하지 않음)에 대한 결과입니다. 단백질-표적 연관성에 대한 명백한 의사 1차 속도 상수 kapp이 각 경우에 대해 표시됩니다. (F) 단백질-표적 연관성에 대한 자가억제의 동역학적 영향. 이 패널은 k app/k app,o 비율을 보여주며, 여기서 k app 및 k app,o는 각각 자동 억제가 있거나 없는 시스템에서 단백질-표적 복합체(PT)의 형성에 대한 명백한 의사 500차 속도 상수를 나타냅니다. 패널 C-F의 경우, 다음의 평형 및 속도 상수가 사용되었다. 해리 상수 : Kd, PT = 1 nM; Kd, PD = 500 nM; KD, XT = 10 μM 및 KD, XD = <> μM. 고유 연관률 상수: k on,PD= k on,PT = k on,XD = k on,XT = <> 8 M−1s−1. 구조적 전환에 대한 속도 상수: k XP = k XTPT = kXDPD = 10 3 s−1; k PX = kXP케이아이; k PTXT = k XTPT K ai K d,PT/K d,XT 및 k PDXD = kXDPD K ai K d,PD/K d,XD입니다. k PTXT 및 k PDXD에 대한 방정식은 상세 균형의 원리에 기초한다 (84). 패널 D-F에 대해, Ptot = 200 nM, Ttot = 10 nM 및 Dtot = 8000 nM이 사용되었고, 여기서 Ptot, Ttot 및 Dtot는 각각 단백질, 표적 및 미끼의 총 농도를 나타낸다.
그림 5B에 표시된 운동 모델은보다 정량적 인 설명을 제공합니다. 이 모델은 구조적 선택(X→P→PT) 및 유도 적합성(X→XT→PT)에 대한 Hammes 등(70)의 모델과 유사합니다. 자가억제에 대한 평 형 상수 Kai는 Kai = [X]eq/[P]eq에 의해 정의되며, 여기서 []eq는 평형 농도를 나타낸다. 카이> 단백질 분자의 대부분이 자가억제될 때 1이다. 억제되지 않은 단백질 (P)은 표적 (T) 또는 미끼 (D)와 안정한 복합체 (PT 또는 PD)를 형성 할 수 있으며, 표적에 대한 친화도는 실질적으로 더 강하며, 이는 해리 상수 (Kd)의 관점에서 Kd, PT << Kd, PD를 의미한다. 자가 억제 단백질 (X)은 CGMD 시뮬레이션에서 볼 수 있듯이 표적 또는 미끼와 약하게 상호 작용하여 일시적인 복합체 (XT 또는 XD)를 형성 할 수 있습니다 (그림 4F). 또한, 정전기 반발력은 D/E 반복을 DNA에서 멀리 방출하고 억제되지 않은 복합체로의 구조적 전이를 유도합니다(그림 4G, 영화 S1 참조).
자동 억제는 표적에 대한 겉보기 친화도를 약화시키며, 이에 대해 겉보기 해리 상수(Kd,app)는 보충 데이터에 설명된 바와 같이 K d,PT(1 + Kai)/(1 + K ai K d,PT/Kd,XT에 의해 주어집니다. 도 5C는 이러한 충격을 자가억제에 대한 평형 상수 Kai의 함수로서 보여준다. 예를 들어, Kai= 50일 때, 자가억제는 겉보기 친화도를 ∼50의 계수만큼 약화시킨다. 자동 억제가 표적 결합 상태의 평형 집단의 급격한 감소를 야기 할 것으로 예상 할 수있다. 그러나 흥미롭게도 시스템이 압도적 인 수의 미끼를 포함하는 경우 단백질-표적 복합체 (PT)의 평형 집단은자가 억제가없는 시스템에서보다자가 억제 시스템에서 약간 더 작을 수 있습니다 (그림 5D). 이 반 직관적 인 효과는 감소 된 친 화성으로 인해 더 적은 양의 단백질이 미끼에 갇히기 때문에 발생합니다.
이 운동 모델을 사용한 시뮬레이션을 통해 우리는 자동 억제가 미끼가 있는 상태에서 단백질-표적 연관 역학에 어떤 영향을 미치는지 조사했습니다. 모델에 포함된 비율 상수는 그림 캡션에 표시되고 비율 방정식은 보충 데이터에 제공됩니다. 조건은 도 1C에 도시된 HMGB2 데이터를 모방하도록 선택되었다. 그림 5E에서 빨간색 곡선은 미끼가 있는 상태에서 자가 억제를 포함하는 시스템에 대한 단백질-표적 연관성에 대한 시간 경과 데이터를 보여주는 반면, 검은색 곡선은 자동 억제가 포함되지 않은 해당 시스템에 대한 데이터를 보여줍니다. 이러한 결과는 그림 2C에 표시된 실험 데이터와 유사하며, D/E 반복을 통한 동적 자동 억제가 있는 경우 더 빠른 표적 검색 역학을 시사합니다. 도 5F는 가속도의 정도(즉, 동적 자기억제의 존재 및 부재 하에서의 표적 탐색에 대한 겉보기 속도 상수 kapp/kapp,o의 비율)를 자기억제에 대한 카이 상수의 함수로서 나타낸다. 이러한 데이터는 동적 자가 억제가 미끼가 있는 상태에서 단백질-표적 결합을 크게 가속화할 수 있음을 예측합니다.
우리의 운동 시뮬레이션은 자동 억제가 유도 된 맞춤 경로가 효율적 일 때 (그리고 경우에만) 단백질-표적 연관성을 가속화 할 수 있음을 시사합니다. 형태 선택 (즉, X→P→PT) 및 유도 적합 (즉, X→XT→PT) 경로의 플럭스는 다음과 같습니다. 보충 그림 S5 보충 데이터에서. 미끼 분자 (D)가있는 시스템에서는 억제되지 않은 상태 (P)의 단백질이 미끼에 쉽게 포획 될 수 있기 때문에 구조적 선택 경로가 효율적이지 않습니다. 중요한 것은 D/E 반복을 통한 자동 억제의 경우 CGMD 시뮬레이션에서 알 수 있듯이 음전하를 띤 IDR과 DNA 사이의 정전기 반발을 통해 유도 피팅이 효율적으로 발생할 수 있다는 것입니다(그림 4G 및 영화 S1).
방정식 (1)은 자가억제에 의한 단백질-표적 결합의 가속화에 대한 유용한 통찰력을 제공한다. 가속에 필요한 조건은 자가 억제 단백질의 고유 회합 속도(kon,XT)가 억제되지 않은 단백질의 고유 회합 속도(kon,PT)에 비해 충분히 빠르다는 것입니다. 그렇지 않으면 k on, XT / kon, PT << 1로 인해 가속 효과가 감소합니다. Antp HD-DERT 단백질에 대한 CGMD 시뮬레이션은 방정식 (1)의 k on, XT / kon, PT 항이 1보다 훨씬 클 수 있음을 보여 주며, 이는 가속 효과에 매우 적합합니다. 또 다른 필요한 조건은 자동 억제 복합체 (XT)에서 억제되지 않은 복합체 (PT)로의 구조적 전이가 XT의 해리에 비해 충분히 빠르다는 것입니다. 그렇지 않으면 k XTPT / (k XTPT + k off, XT) << 1로 인해 가속도가 감소합니다. CGMD로부터 계산된 k XTPT/(k XTPT + koff,XT)의 값은 0개 잔기의 DERT에 대해 9.5에서 0개의 잔기를 갖는 DERT의 경우 02.16로 변한다. 따라서 수식 1과 함께 CGMD 데이터는 가속 효과에 대한 최적의 길이가 있는 이유를 설명합니다(그림 3). 대부분의 경우 Antp HD-DERT16의 느린 표적 연결은 XT에서 PT로의 느린 구조적 전환 때문입니다.
토론
우리의 현재 연구는 적절한 길이의 D / E 반복을 통한 동적 자동 억제가 표적 DNA 검색 과정을 가속화 할 수 있음을 보여줍니다. 이것은 분자 스위치에서 '꺼짐'상태를 만드는 메커니즘으로서의 자동 억제의 전통적인 개념과 근본적으로 다릅니다 (21-23). 가속 효과는 D/E 반복이 수많은 미끼가 포함된 시스템에서 검색 프로세스를 방해하는 미끼와의 연관성을 억제하기 때문에 발생합니다. 즉, D/E 반복에 의한 자동 억제는 단백질이 미끼의 주의를 산만하게 하는 것을 피하여 표적을 더 빠르게 포착할 수 있도록 합니다.
당사의 거친 시뮬레이션과 키네틱 모델은 D/E 반복이 어떻게 표적 검색 동역학을 가속화할 수 있는지에 대한 기계론적 통찰력을 제공합니다. 이 메커니즘은 검색 효율성과 결합 친화도 사이의 트레이드 오프 개념과 유사합니다(68). 그러나 선호도를 줄이는 것만으로는 검색 속도를 높이기에 충분하지 않습니다. 가속을 위한 핵심 요구 사항은 DNA를 만났을 때 단백질이 DNA와 상호 작용하는 동안 자가 억제 형태에서 억제되지 않은 형태로 효율적으로 전환되어야 한다는 것입니다. DNA의 정전기 반발력은 구조적 변화를 유도하는 것으로 보이며, 영화 S1에서 볼 수 있듯이 음전하를 띤 D/E 반복을 DNA 결합 도메인에서 멀어지게 합니다. 단백질-표적 회합의 생물학적으로 의미 있는 가속을 야기하는 자가억제를 위해서는, 평형 상수 Kai의 최적 범위가 존재한다. 자동 억제가 너무 약하면 미끼에 의한 트래핑으로 인해 가속 효과가 너무 작아집니다. 자가 억제가 너무 강하면 단백질-표적 복합체의 평형 집단이 너무 낮아 단백질의 기능이 저하됩니다. 그림 5D–F에 사용된 시스템의 경우, Kai가 ∼10–10일 때2, 단백질-표적 복합체의 평형 집단을 심각하게 감소시키지 않으면서 상당한 가속이 달성될 수 있다. HMGB1 (13) 및 Antp HD-DERT11 단백질에 대한 Kai 상수가 이 범위에 속한다. 두 시스템 모두에서 DNA에 대한 친화도는 결합 속도의 약 100배 증가와 결합된 D/E 반복을 통한 자동 억제에 의해 약 10배 감소했습니다.
수백 개의 단백질이 각 포유류 프로테옴에서 D/E 반복을 가지고 있습니다(20). 예를 들어, 268개의 인간 단백질과 275개의 마우스 단백질은 10개 이상의 연속 잔기의 D/E 반복을 포함합니다. 그러나 D/E 반복의 기능에 대해서는 알려진 바가 거의 없습니다. 일부 D/E 반복은 자동 억제(5-13)를 겪는 것으로 입증되었으며, 다른 반복은 보호자와 같은 활동에서 역할을 하는 것으로 제안되었습니다(14,71). HMGB1의 경우, 뉴 클레오 솜 리모델링에서 D / E 반복의 역할도 제안되었다 (72). 흥미롭게도 D / E 반복을 포함하는 모든 단백질의 ∼50 %는 DNA / RNA 결합 단백질입니다 (20). 우리의 현재 연구에서 입증 된 바와 같이, 이러한 DNA / RNA 결합 단백질은 D / E 반복을 사용하여 수십억 개의 염기쌍을 포함하는 게놈에서 표적을 효율적으로 찾을 수 있습니다. 특정 서열을 인식하는 전사 인자의 경우 포유류 게놈에는 수백만 개의 미끼가 있는 반면 기능적 표적은 훨씬 적습니다(30,31). D/E 반복에 의한 동적 자동 억제는 미끼에 의한 격리 위험을 줄일 수 있습니다. RNA 결합 단백질은 RNA가 세포의 DNA보다 훨씬 풍부하기 때문에 비슷한 상황에 직면 할 가능성이 높습니다 (73). D/E 반복은 고등 진핵생물에서 더 자주 발견됩니다(20). 이것은 더 큰 게놈이 더 많은 미끼를 부과하기 때문에 고등 진핵생물에서 DNA/RNA 결합 단백질에 의한 표적 검색의 촉진제로서 D/E 반복의 중요성과 관련이 있을 수 있습니다.
D/E 반복을 통한 자동 억제는 대상 검색 프로세스를 가속화하는 데 매우 적합한 것으로 보입니다. DNA/RNA 결합 단백질의 다른 유형의 자가억제도 표적 검색 과정을 가속화하는지 여부는 여전히 조사되어야 합니다. 예를 들어, ADR2, C / EBPβ, Ets-1, ETV6, GCN2, Hfq, p53 및 U2AF2 (25,26,28,74-80)의자가 억제는 이와 관련하여 잠재적으로 흥미로운 조사 대상입니다. 가속은 표적(PT)과의 억제되지 않은 복합체로의 효율적인 유도 적합 전이를 유도하는 일시적인 복합체 (도 5B에 도시된 동역학 모델에서 XT)를 형성하는 자가억제 단백질의 능력을 필요로 한다. 다른 IDR을 통한 자동 금지는 D/E 반복과 유사한 동작을 나타낼 수 있습니다.
우리의 연구는 D/E 반복을 통한 단백질 공학의 타당성을 보여줍니다. 원하는 결과를 얻으려면 Antp HD-DERT 구조의 경우와 같이 D/E 반복 및 링커의 길이를 최적화해야 할 수 있습니다(그림 3). D/E 반복 꼬리가 너무 길면 자동 억제가 너무 강하여 대상 검색 프로세스가 가속화되지 않을 수 있습니다. D/E 반복 꼬리가 너무 짧으면 자동 억제가 충분하지 않을 수 있습니다. 링커는 또한 그의 길이 및 유연성이 기능 도메인 (81,82)에 대한 억제 세그먼트의 유효 농도에 영향을 미치기 때문에 최적화되어야 할 수 있다. 단백질 구조체에 D/E 반복을 추가하는 것은 비교적 쉬우며 단백질 엔지니어링에 유용한 도구가 될 수 있습니다.
결론적으로, 우리는 특정 조건에서 동적 자가 억제가 미끼와 관련된 시스템에서 단백질-표적 결합을 가속화할 수 있음을 입증했습니다. HMGB1에서 관찰된 바와 같이, D/E 반복을 포함하는 천연 단백질은 이 메커니즘을 사용하여 수많은 미끼가 포함된 환경에서 표적과 효율적으로 결합할 수 있습니다. 이 메커니즘은 Antp HD-DERT 단백질에 대해 입증된 바와 같이 단백질 엔지니어링을 통해 다른 시스템에서 구현될 수 있습니다. D/E 반복을 통한 이러한 인공 자가억제는 설계/설계된 단백질의 동역학적 특성을 향상시키는 유용한 도구가 될 수 있습니다. 또한 표적 검색을 가속화하기 위해 자동 억제에 필요한 조건에 대한 통찰력을 얻었습니다. 이러한 조건을 충족하기에 적합한 적절한 길이의 D/E 반복을 통해 단백질은 미끼의 주의를 산만하게 하는 것을 방지하고 표적과 빠르게 결합할 수 있습니다.
데이터 가용성
모든 데이터는 논문과 보충 데이터에서 사용할 수 있습니다.
보충 데이터
Negatively charged, intrinsically disordered regions can accelerate target search by DNA-binding proteins
Abstract. In eukaryotes, many DNA/RNA-binding proteins possess intrinsically disordered regions (IDRs) with large negative charge, some of which involve a conse
academic.oup.com
Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation - PubMed
Allosteric regulatory processes are implicated at all levels of biological function. Recent advances in our understanding of the diverse and functionally significant class of intrinsically disordered proteins have identified a multitude of ways in which di
pubmed.ncbi.nlm.nih.gov
search
Modify your search 1-20 of 206697 Sort by Sort Order Select Relevance Date – Newest First Date – Oldest First Title - A to Z Title - Z to A Published: 13 February 2023 Figure 1. Examples of D/E repeats in human DNA-binding proteins. Autoinhibition by t
academic.oup.com

'생명공학' 카테고리의 다른 글
| 고통 저감 유전자 드라이브 (0) | 2023.02.28 |
|---|---|
| 유전체 편집: genome editing (0) | 2023.02.28 |
| NCBI GEO 유전체학 데이터 (0) | 2023.02.14 |
| NCBI 생명 공학 정보 센터 (0) | 2023.02.14 |
| 게놈 데이터 포털 (0) | 2023.01.11 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}